医薬品リスク管理計画について
はじめに
「医薬品リスク管理計画」(Risk Management
Plan:以下、RMP)は、個々の医薬品について安全性上の検討課題を特定し、使用成績調査、市販直後調査等による調査・情報収集や、医療関係者の皆様への追加の情報提供などの医薬品のリスクを低減するための取り組みを、医薬品ごとに文書化したものです。平成25年4月1日以降に製造販売承認申請した新医薬品とバイオ後続品からRMPの策定が求められています。
医療関係者の皆様には、医薬品リスク管理計画書として公表されるRMPのご活用ととともに、医薬品のリスク最小化活動及び安全性監視活動の実施にご理解とご協力をお願い申し上げます。
「医薬品リスク管理計画」(RMP)の概要
医薬品は、有効性とともに一定のリスク(副作用)を伴うものであり、リスクをゼロにすることはできませんが、これを可能な限り低減するための方策を講じ、適切に管理していくことが重要です。
医薬品の安全性を確保するためには、開発の段階から承認審査を経て製造販売後に至るまで、常に医薬品のリスクを適切に管理する方策を検討することが重要です。今回導入されたRMPは、現在行われているこれらの取り組みを医薬品ごとに文書化し、医療関係者の皆様、行政、製薬企業など医薬品の適正使用に関わるすべての関係者で共有できるようにすることで、市販後安全対策の一層の充実強化を図ることを目的とするものです。
RMPは、基本的に3つの要素(「安全性検討事項」「医薬品安全性監視計画」「リスク最小化計画」)から構成されます。また、これらに加え、必要に応じて有効性に関する調査・試験の計画を作成することが求められます。これら計画の全体を取りまとめ文書化したものがRMPです。
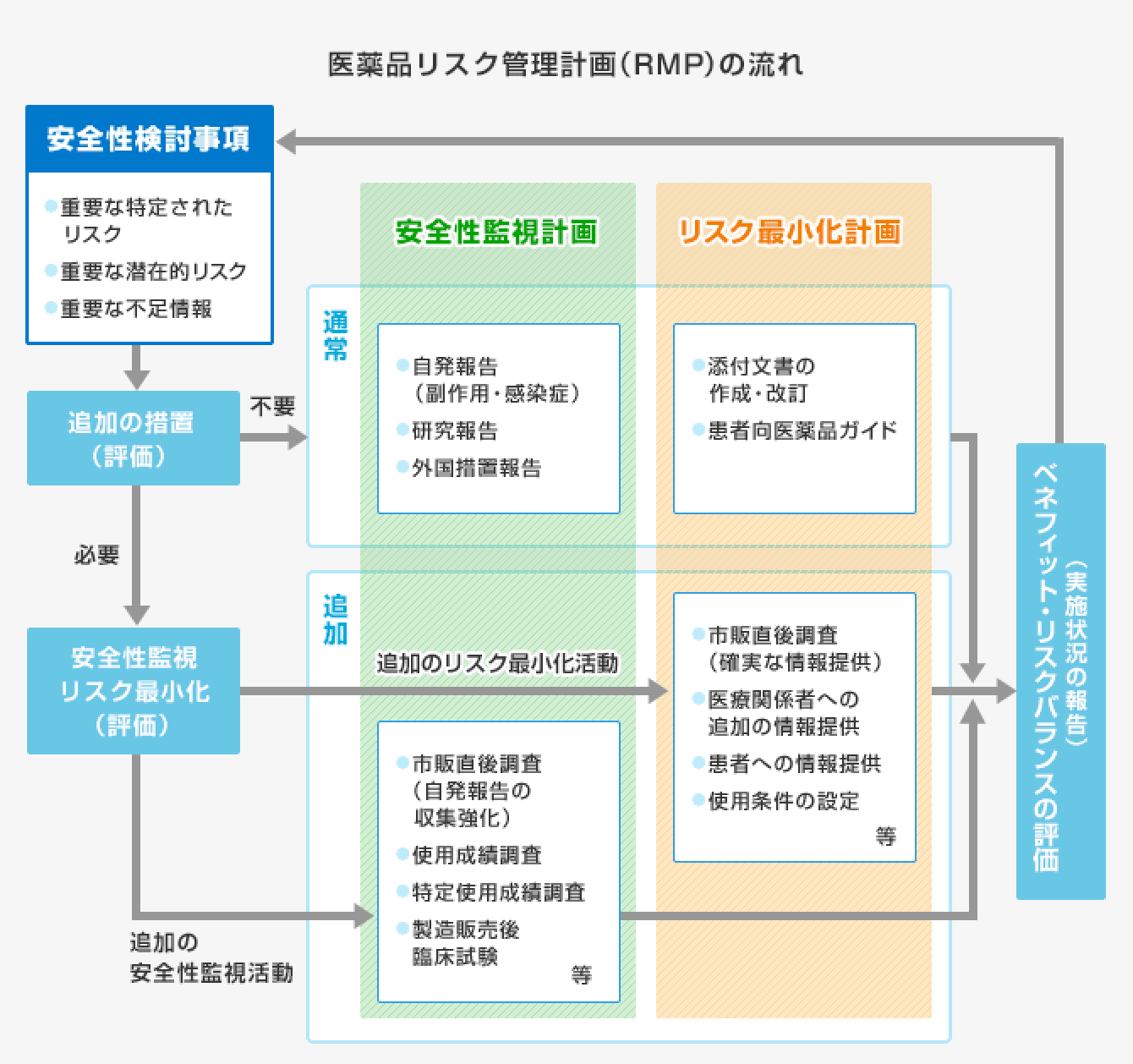
RMPの3つの要素
安全性検討事項
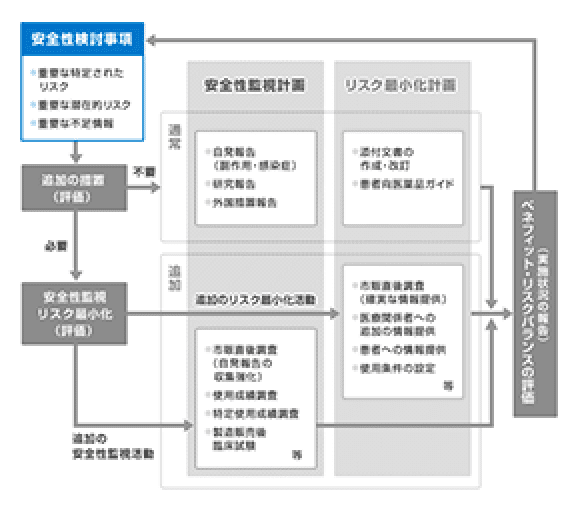
「安全性検討事項」とは、開発段階で得られた情報や市販後の副作用報告などから明らかとなったリスクのうち、医薬品のベネフィット・リスクバランスに影響を及ぼしうる、又は保健衛生上の危害の発生・拡大のおそれがあるような重要な事項のことです。「重要な特定されたリスク」、「重要な潜在的リスク」、「重要な不足情報」の3つのリスク・情報として特定します。
- 臨床試験において本剤群で有意に発現している副作用
- 多くの自発報告があり、時間的関連性等から因果関係が示唆される副作用
- 薬理作用等から予測されるが、臨床的には確認されていない副作用
- 同種同効薬で認められている副作用
- 治験対象から除外されているが実地医療では高頻度で使用が想定される患者集団(高齢者、腎機能障害患者、肝機能障害患者、妊婦、小児など)における安全性情報
医薬品安全性監視計画
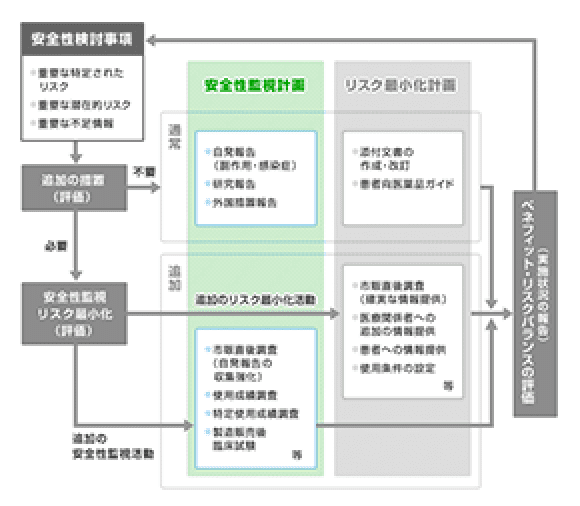
「医薬品安全性監視計画」とは、特定された「安全性検討事項」を踏まえて、市販後に情報を収集する目的で実施される調査・試験の計画です。“医薬品安全性監視活動”には「通常の医薬品安全性監視活動」と「追加の医薬品安全性監視活動」があります。
副作用症例や文献情報等の収集
市販直後調査、使用成績調査、特定使用成績調査、製造販売後臨床試験、薬剤疫学研究などによる情報収集
リスク最小化計画
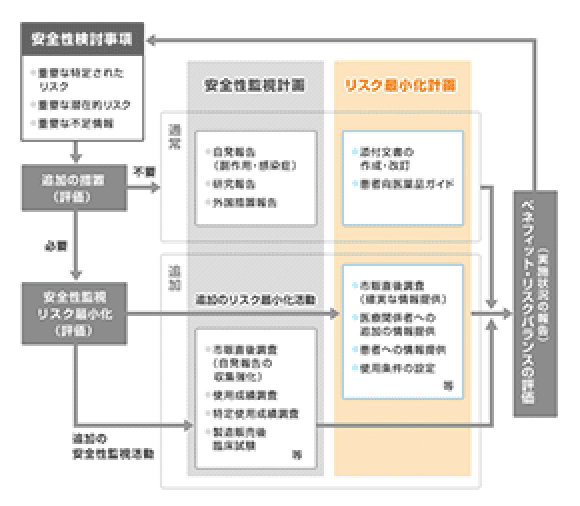
「リスク最小化計画」とは、開発段階で得られた情報や市販後の副作用報告などから明らかとなったリスクを最小に抑えるための安全対策の計画です。
“リスク最小化活動”には「通常のリスク最小化活動」と「追加のリスク最小化活動」があります。医薬品によっては重大なリスクの更なる低減のために、市販直後調査による医療関係者への頻繁な注意喚起や重要な注意を要する医薬品について適正使用を周知するための資材の配布を行う場合や、登録した医師のみ処方を可能とする、患者にインフォームドコンセントを得た上で投与するといった条件の設定などが「追加のリスク最小化活動」です。
電子添文の作成・改訂、患者向医薬品ガイド
市販直後調査による情報提供、適正使用のための資材の配布、使用条件の設定など
RMPは一度策定すれば終わりというものではなく、市販後に得られた新たな安全性・有効性の情報に基づき常に見直しを行う必要があります。例えば、新たな副作用が判明した場合など安全性検討事項の内容に変更があった時、計画に基づき実施した調査又は試験が終了して新たな知見が得られた時などが挙げられます。
このようにRMPは医薬品のライフサイクルの中で計画の策定、実施、評価、見直しが継続して行われていくことになります。
医療関係者の皆様へのお願い
医薬品をより有効かつ安全に使用するために
製薬企業が行う『医薬品リスク管理計画(RMP)』について
先生方のご理解とご協力をお願い申し上げます。
- 平成25年4月1日以降に承認申請した新医薬品及びバイオ後続品では、承認申請時にRMPの案を提出し、承認審査における検討を経て、承認後に最終的なものを提出することになります。
- また、平成25年4月1日以降に新たな安全性の懸念が判明した既承認の品目についても新たにその提出が求められています。
- RMPは医薬品リスク管理計画書として、(独)医薬品医療機器総合機構(PMDA)のホームページで公表されます。
医療関係者の皆様におかれましては、RMPの内容を把握していただき、それぞれの医薬品に現時点でどのようなリスクがあり、それに対してどのような安全対策が実施されているかをご理解いただき、適正使用に活用していただきますようお願い申し上げます。また、市販後の調査・試験の円滑な実施には、医療関係者の皆様のご理解とご協力が必要であり、どのようなリスクについてどのような調査・試験が実施されているかをご理解いただき、積極的に参画していただきますようご協力をお願い申し上げます。
(参考文献)
- 1)
医薬品安全性監視の計画について
(平成17年9月16日付薬食審査発0916001号・薬食安発0916001号厚生労働省医薬食品局審査管理課長・安全対策課長連名通知)
(日米EU医薬品規制調和国際会議(ICH)情報ホームページ) - 2)
医薬品リスク管理計画指針について
(平成24年4月11日付薬食安発0411第1号・薬食審査発0411第2号厚生労働省医薬食品局安全対策課長・審査管理課長連名通知)
(医薬品医療機器情報提供ホームページ) - 3)
医薬品リスク管理計画の策定について
(平成24年4月26日付薬食審査発0426第2号・薬食安発0426第1号厚生労働省医薬食品局審査管理課長・安全対策課長連名通知)
(医薬品医療機器情報提供ホームページ)
編集 日本製薬工業協会 医薬品評価委員会 PMS部会 改変 2013年8月